ตำรายาของประเทศไทย
Thai Pharmacopoeia
สำนักยาและวัตถุเสพติด กรมวิทยาศาสตร์การแพทย์ กระทรวงสาธารณสุข
Bureau of Drug and Narcotic, Department of Medical Sciences, Ministry of Public Healthสำนักยาและวัตถุเสพติด กรมวิทยาศาสตร์การแพทย์ กระทรวงสาธารณสุข
Bureau of Drug and Narcotic, Department of Medical Sciences, Ministry of Public Health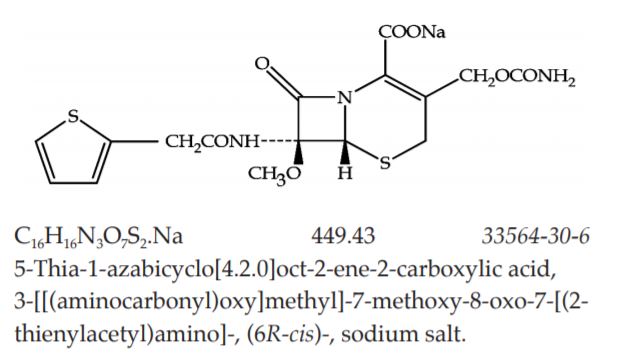
Category Antibacterial (second-generation cephalosporin).
Cefoxitin Sodium contains the equivalent of not less than 95.0 per cent and not more than 102.0 per cent of C16H17N3O7S2, calculated on the anhydrous basis.
Description White to off-white, granules or powder; odour, slight and characteristic.
Solubility Very soluble in water; soluble in methanol; sparingly soluble in dimethylformamide; slightly soluble in acetone; insoluble in chloroform and in ether.
Contra-indication; Additional information See under Cephalexin, p. 58.
Warning
1. Pain, tenderness and induration have been reported with intramuscular administration and thrombophlebitis may occur with intravenous administration.
2. Concurrent use of nephrotoxic agents may increase the risk of nephrotoxicity.
3. It may cause transient increase in serum aspartate transminase (AST), alanine transaminase (ALT), lactate dehydrogenase (LDH) and alkaline phosphatase concentrations and jaundice.
See also under Cephalexin, p. 58.
Packaging and storage Cefoxitin Sodium shall be kept in tightly closed containers and stored at a temperature between 2º and 8º.
Identification
A. The infrared absorption spectrum is concordant with the spectrum obtained from Cefoxitin RS (Appendix 2.1) or with the reference spectrum of Cefoxitin.
B. The retention time of the major peak in the chromatogram of the Assay preparation corresponds to that in the chromatogram of the Standard preparation, as obtained in the Assay.
C. The ultraviolet absorption spectrum, when observed between 220 and 400 nm, of a 0.002 per cent w/v solution in phosphate buffer prepared by dissolving 1.0 g of potassium dihydrogenphosphate and 1.8 g of anhydrous sodium hydrogenphosphate in water to make 1000 ml, exhibits maxima at the same wavelengths as those of a similar solution of Cefoxitin Sodium RS, concomitantly measured (Appendix 2.2).
D. It yields the reactions characteristic of sodium salts (Appendix 5.1).
Crystallinity It is crystalline (Method I, Appendix 4.14).
pH 4.2 to 7.0, in a 10.0 per cent w/v solution (Appendix 4.11).
Specific rotation +206º to +214º, calculated on the anhydrous basis, determined in a solution prepared by dissolving 250 mg in methanol and diluting to 25.0 ml with the same solvent (Appendix 4.8).
Water Not more than 1.0 per cent w/w (Karl Fischer Method, Appendix 4.12). Use 500 mg.
Absorbance Dissolve 100.0 mg in water and dilute to 100.0 ml with the same solvent. Dilute 2.0 ml of the solution to 100.0 ml with a 4.2 per cent w/v solution of sodium hydrogencarbonate. The light absorption spectrum of the diluted solution, when observed between 220 nm and 350 nm, exhibits a maximum at about 236 nm and a broad absorption maximum at about 262 nm; the specific absorbance at this broad maximum is 190 to 210, calculated on the anhydrous basis (Appendix 2.2).
Related substances Carry out the test as described in
the “High-pressure Liquid Chromatography” (Appendix
3.5). Prepare the solutions immediately before use.
Mobile phase
Mobile phase A Use water adjusted to pH 2.7 with anhydrous formic acid.
Mobile phase B Use acetonitrile.
Solution A Dilute 20 ml of a 34.8 g/l solution of dipotassium hydrogenphosphate adjusted to pH 6.8 with phosphoric acid to 1000 ml with water.
Test solution Dissolve 50.0 mg of the test substance in Solution A and dilute to 10.0 ml with the same solution.
Reference solution (a) Dilute 1.0 ml of Test solution to 100.0 ml with Solution A.
Reference solution (b) To 1.0 ml of Test solution, add 7.0 ml of water and 2.0 ml of methanol. Add 25 mg of sodium carbonate, stir for 10 minutes at room temperature, and then heat in a water-bath at 70º for 30 minutes. Allow to cool. Add 3 drops of glacial acetic acid and 1 ml of Test solution and mix.
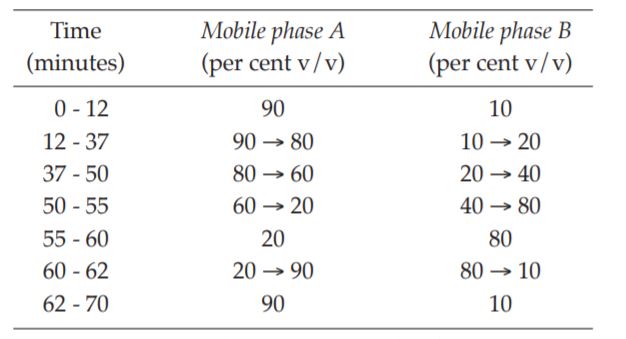
Chromatographic system The chromatographic procedure may be carried out using (a) a stainless steel column (25 cm × 4.6 mm) packed with phenylsilyl silica gel for chromatography or ceramic microparticles (5 μm) with a specific surface area of 300 m2/g and a pore size of 7 nm, (b) Mobile phase at a flow rate of about 1 ml per minute, and (c) an ultraviolet photometer set at 235 nm. The step gradient of mobile phases is as follows:
To determine the suitability of the chromatographic system, chromatograph Reference solution (b), and record the peak responses as directed under Procedure: theresolution factor between the two principal peaks is not less than 5.0.
Procedure Separately inject equal volumes (about 50 μl) of Reference solutions (a) and (b), and Test solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks. The relative retention times are about 0.82 for impurity A, about 1.16 for impurity B, about 1.27 for impurity C, about 1.31 for impurity D and 1.0 for cefoxitin (retention time about 34 minutes).
In the chromatogram obtained from Test solution, determine the percentage content of related substances by using the area of the principal peak in the chromatogram obtained from Reference solution (a) (1.0 per cent) as a comparison area.
Limits
Disregard limit Not more than 0.05 times the comparison area (0.05 per cent).
Any impurity Not more than 0.5 times the comparison area (0.5 per cent).
Total Not more than 4 times the comparison area (4.0 per cent).
Assay Carry out the determination as described in the “High-pressure Liquid Chromatography” (Appendix 3.5).
Mobile phase Prepare a mixture of 84 volumes of water, 16 volumes of acetonitrile and 1 volume of glacial acetic acid. Make adjustments if necessary.
Phosphate buffer Dissolve 1.0 g of potassium dihydrogenphosphate and 1.8 g of sodium hydrogenphosphate in 900 ml of water, adjust with phosphoric acid or 10 M sodium hydroxide to a pH of 7.1±0.1, dilute with water to make 1000 ml, and mix.
Standard preparation Dissolve an accurately weighed quantity of Cefoxitin RS in Phosphate buffer pH 7.1 to obtain a solution having a known concentration of about 300 μg per ml. (Sonicate, if necessary, to dissolve the sample. Use this solution within 5 hours.)
Assay preparation Transfer about 150 mg of Cefoxitin Sodium, accurately weighed, to a 500-ml volumetric flask, dissolve in and dilute with Phosphate buffer pH 7.1 to volume, and mix. (Sonicate, if necessary, to dissolve the specimen. Use this solution within 5 hours.)
Chromatographic system The chromatographic procedure may be carried out using (a) a stainless steel column (30 cm × 3.9 mm) packed with octadecylsilane chemically bonded to porous silica or ceramic microparticles (5 to 10 μm), (b) Mobile phase at a flow rate of about 1 ml per minute, and (c) an ultraviolet photometer set at 254 nm.
To determine the suitability of the chromatographic system, chromatograph Standard preparation, and record the peak responses as directed under Procedure: the relative standard deviation for replicate injections is not more than 1.0 per cent and the symmetry factor for the cefoxitin peak is not more than 1.5.
Procedure Separately inject equal volumes (about 10 μl) of Standard preparation and Assay preparation into the chromatograph, record the chromatograms, and measure the responses for the major peaks.
Calculation Calculate the content of C16H17N3O7S2 in the Cefoxitin Sodium taken, using the declared content of C16H17N3O7S2 in Cefoxitin RS.
Other requirements Cefoxitin Sodium intended for parenteral administration complies with the following additional requirements.
Bacterial endotoxins When test as described the “Test for Bacterial Endotoxins” (Appendix 8.5), it contains not more than 0.13 Endotoxin Unit per mg of cefoxitin.
Sterility Complies with the “Sterility Test” (Method I, Appendix 10.1).