ตำรายาของประเทศไทย
Thai Pharmacopoeia
สำนักยาและวัตถุเสพติด กรมวิทยาศาสตร์การแพทย์ กระทรวงสาธารณสุข
Bureau of Drug and Narcotic, Department of Medical Sciences, Ministry of Public Healthสำนักยาและวัตถุเสพติด กรมวิทยาศาสตร์การแพทย์ กระทรวงสาธารณสุข
Bureau of Drug and Narcotic, Department of Medical Sciences, Ministry of Public Health5.2 LIMIT TESTS
ALUMINIUM
Extract the prescribed solution with successive portions of 20, 20 and 10 ml of a 0.5 per cent w/v solution of 8-quinolinol in chloroform and dilute the combined extracts to 50.0 ml with chloroform. Unless otherwise stated in the monograph, use as the blank solution a mixture of 10 ml of acetate buffer pH 6.0 and 100 ml of water treated in the same manner and as the standard solution a mixture of 2.0 ml of aluminium standard solution (2 ppm Al), 10 ml of acetate buffer pH 6.0 and 98 ml of water treated in the same manner. Measure the fluorescence of the test solution and of the standard solution (Appendix 2.4), using an excitation wavelength of 392 nm and a secondary filter with a transmission band centred at 518 nm, or a monochromator set to transmit at this wavelength, and setting the instrument to zero with the blank solution in each case. The fluorescence of the test solution is not greater than that of the standard solution.
HEAVY METALS
This test is provided to demonstrate that the content of metallic impurities that are coloured by sulfide ion, under the specified test conditions, does not exceed the heavy metals limit specified in the individual monograph in terms of parts per million (ppm), or, when the limit exceeds 100 ppm, in terms of a percentage (by weight), of lead in the test substance, as determined by concomitant visual comparison with a control prepared from a lead standard solution.
Method I is used for substances that yield clear, colourless preparations under the specified test conditions. Method II is used for substances that do not yield clear, colourless preparations under the test conditions specified for Method I, or for substances that, by virtue of their complex nature, interfere with the precipitation of metals by sulfide ion, or for fixed and volatile oils.
Reagent
Thioacetamide reagent To 1.0 ml of a 4 per cent w/v solution of thioacetamide, add 5 ml of a mixture of 15 ml of 1 M sodium hydroxide, 5 ml of water and 20 ml of glycerol. Heat on a water-bath for 20 seconds, cool, and use immediately.
Method I
Standard Preparation Transfer 10.0 ml of lead standard solution (1 ppm Pb or 2 ppm Pb), as prescribed, to a 50-ml comparison tube, add 2.0 ml of the solution prepared from the test as described in the individual monograph, add 2 ml of acetate buffer pH 3.5, and mix.
Test Preparation
Into a 50-ml comparison tube place 12.0 ml of the solution prepared for the test as directed in the individual monograph, add 2 ml of acetate buffer pH 3.5, and mix.
Procedure To each of the tubes containing the standard preparation and the test preparation, add 1.2 ml of thioacetamide reagent, mix, allow to stand for 2 minutes, and view downward over a white surface: the colour of the solution from the test preparation is not darker than that of the solution from the standard preparation.
Method II
Standard Preparation Transfer 10.0 ml of lead standard solution (1 ppm Pb or 2 ppm Pb), as prescribed, to a 50-ml comparison tube, add 2.0 ml of solution A (see under test preparation below). Add 2 ml of acetate buffer pH 3.5 and mix.
Test Preparation Accurately weigh the prescribed quantity of the substance being examined, in a suitable crucible, preferably made of silica, and carefully ignite at a low temperature until the contents are thoroughly charred. The crucible may be loosely covered with a lid during the charring. Add to the contents of the crucible 2 ml of nitric acid and 5 drops of sulfuric acid, and heat cautiously until white fumes are no longer evolved. Ignite, preferably in a muffle furnace, at 500º to 600º, until the carbon is completely burned off. Cool, add 4 ml of 6 M hydrochloric acid, cover, digest on a waterbath for 15 minutes, uncover, and slowly evaporate on a water-bath to dryness. Moisten the residue with 1 drop of hydrochloric acid, add 10 ml of hot water, and digest for 2 minutes. Add 0.1 ml of diluted phenolphthalein TS (1 in 10) and 6 M ammonia dropwise, until a pink colour is obtained. Cool, add glacial acetic acid until the solution is decolorized and add a further 0.5 ml. Filter if necessary, and dilute the solution to 20.0 ml with water (solution A). To 12.0 ml of solution A add 2 ml of acetate buffer pH 3.5 and mix.
Procedure To each of the tubes containing the standard preparation and the test preparation, add 1.2 ml of thioacetamide reagent, mix, allow to stand for 2 minutes, and view downward over a white surface: the colour of the solution from the test preparation is not darker than that of the solution from the standard preparation.
Method III
Standard Preparation Prepare a standard solution similar to solution A as described under test preparation, but using the prescribed volume of lead standard solution (10 ppm Pb) in place of the test substance. Transfer 10.0 ml of the resulting solution to a 50-ml comparison tube and add 2.0 ml of solution A. Add 2 ml of acetate buffer pH 3.5 and mix.
Test Preparation Place the prescribed quantity of the test substance in a silica crucible with 4 ml of a 25 per cent w/v solution of magnesium sulfate in 1 M sulfuric acid. Mix using a fine glass rod, and heat cautiously. If the mixture is liquid, evaporate gently to dryness on a water-bath. Progressively heat to ignition, not allowing the temperature to exceed 800º, and continue heating until a white or greyish residue is obtained. Allow to cool, moisten the residue with a few drops of 1 M sulfuric acid, evaporate, ignite again, and allow to cool. The total period of ignition should not exceed 2 hours. Dissolve the residue using two 5-ml portions of 2 M hydrochloric acid. Add 0.1 ml of dilute phenolphthalein TS and 13.5 M ammonia until a pink colour is obtained. Cool, add glacial acetic acid until the solution is decolorized and add a further 0.5 ml. Filter if necessary, and dilute the solution with water to 20.0 ml (solution A). To 12.0 ml of solution A, add 2 ml of acetate buffer pH 3.5 and mix.
Procedure To each of the tubes containing the standard preparation and the test preparation, add 1.2 ml of thioacetamide reagent, mix, allow to stand for 2 minutes, and view downward over a white surface: the colour of the solution from the test preparation is not darker than that of the solution from the standard preparation.
AMMONIUM
Dissolve the prescribed quantity of the test substance in 14 ml of water in a test-tube, if necessary make alkaline with 2 M sodium hydroxide and dilute to 15.0 ml with water. Add 0.3 ml of alkaline mercuric-potassium iodide TS, stopper the tube, mix, and allow to stand for 5 minutes. Any yellow colour produced is not more intense than that obtained by treating a mixture of 10.0 ml of ammonium standard solution (1 ppm NH4) and 5.0 ml of water in the same manner, unless otherwise specified.
ARSENIC
This photometric procedure, based upon the reaction between silver diethyldithiocarbamate and arsine, is provided to demonstrate that the content of arsenic does not exceed the limit given in the individual monograph.
Apparatus
The apparatus (see illustration) consists of an arsine generator (a) fitted with a scrubber unit (c) and an absorber tube (e) with standard-taper or ground glass ball-and-socket joints (b and d) between the units. However, any other suitable apparatus, embodying the principle of the assembly described and illustrated, may be used.
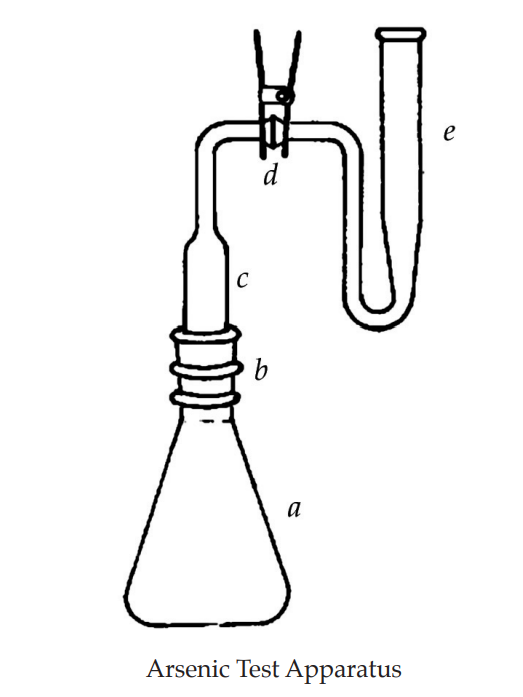
Transfer 3.0 ml of arsenic standard solution (1 ppm As) to the generator flask (a) and add water to make 35 ml.
If the test preparation was prepared as directed in the general procedure for organic compounds, mix the portion of arsenic standard solution (1 ppm As) with 2 ml of sulfuric acid and the total amount of hydrogen peroxide TS (100 volumes) used in the oxidation of the test specimen. Then heat the mixture to strong fuming. Cool, add cautiously 10 ml of water, again evaporate to strong fuming, and cool. Dilute with water to 35 ml.
Test Preparation
|
Caution Some substances may react with explosive violence when digested with hydrogen peroxide. Exercise safety precautions at all times. |
(Note If halogen-containing compounds are present, use a lower temperature while heating the substance to be examined with sulfuric acid. Avoid boiling the mixture and add the hydrogen peroxide with caution, before charring begins, to prevent loss of trivalent arsenic.)
Test preparations of inorganic compounds are prepared as directed in the individual monograph.
Test preparations of organic compounds are prepared according to the following general procedure, unless otherwise directed in the individual monograph.
Transfer the prescribed quantity of the test substance to a 125-ml Kjeldahl flask. Add 5 ml of sulfuric acid and a few glass beads, and digest in a fume hood, preferably on a hot plate, at a temperature not exceeding 120º until charring begins. (Additional sulfuric acid may be necessary to wet some specimens completely, but the total volume added should not exceed 10 ml.) After the test substance has been initially decomposed by the acid, cautiously add, dropwise, hydrogen peroxide TS (100 volumes), allowing the reaction to subside and again heating between drops. Add the first few drops very slowly with sufficient mixing, in order to prevent a rapid reaction. Discontinue heating if foaming becomes excessive. When the reaction has abated, heat cautiously, rotating the flask occasionally to prevent the specimen from caking on glass exposed to the heating unit. Maintain oxidizing conditions at all time during the digestion by adding small quantities of the hydrogen peroxide solution whenever the mixture turns brown or darkens. Continue the digestion until the organic matter is destroyed. Gradually raise the temperature of the hot plate to between 250º and 300º, until fumes of sulfur trioxide are copiously evolved and the solution becomes colourless or retains only a light straw colour. Cool, add cautiously 10 ml of water, mix, and again evaporate to strong fuming. Repeat this procedure, if necessary, to remove any trace of hydrogen peroxide. Cool, add cautiously 10 ml of water, and transfer quantitatively to the generator flask. Dilute with water to 35 ml.
Procedure
To each of the generator flasks containing the standard preparation and the test preparation, add 20 ml of diluted sulfuric acid (1 in 5), 2 ml of potassium iodide TS (16.5 per cent w/v), and 0.5 ml of stronger acid tin(II) chloride TS and mix. Allow to stand at room temperature for 30 minutes. Pack each scrubber unit (c) with two pledgets of cotton that have been soaked in saturated lead acetate solution, freed from excess solution by expression, and dried in vacuum at room temperature, leaving a small space between the two pledgets. Lubricate the joints (b and d) with a suitable stopcock grease (designed for use with organic solvents), and connect each scrubber unit to its absorber tube (e). Transfer 3.0 ml of silver diethyldithiocarbamate TS to each absorber tube. Alternatively, a larger accurately measured volume of the silver diethyldithiocarbamate TS may be employed, provided that the same volume is used for the control test and that the apparatus will accommodate the larger volume. Add 3.0 g of granulated zinc to the mixture in each flask and immediately connect the assembled scrubber unit. Place the generator flasks in a water-bath, maintained at a temperature of 25º±3º, and allow the evolution of hydrogen and the colour development to proceed for 45 minutes, swirling the flasks gently at 10-minute intervals. (If necessary, to obtain a more uniform rate of gas evolution, 1 ml of 2-propanol may be added to each generator.) Disconnect each absorber tube from the generator and scrubber units, and transfer the absorbing solutions to the 1-cm absorption cells. Determine the absorbances of the solutions at the wavelength of maximum absorbance between 535 and 540 nm, with a suitable spectrophotometer, using silver diethyldithiocarbamate TS as the blank. The absorbance due to any red colour from the Test Preparation does not exceed that produced by the Standard preparation.
Interfering Chemicals
Metals or salts of metals such as chromium, cobalt, copper, mercury, molybdenum, nickel, palladium, and silver may interfere with the evolution of arsine. Antimony, which forms stibine, produces a positive interference in the colour development with silver diethyldithiocarbamate TS; when the presence of antimony is suspected, the red colours produced in the two silver diethyldithiocarbamate solutions may be compared at the wavelength of maximum absorbance between 535 and 540 nm, since at this wavelength the interference due to stibine is negligible.
CALCIUM
The solutions used for this test should be prepared with distilled water. To 0.20 ml of ethanolic calcium standard solution (100 ppm Ca) add 1.0 ml of a 4.0 per cent w/v solution of ammonium oxalate. After 1 minute add 1.0 ml of 2 M acetic acid and 15.0 ml of the test solution prepared as directed in the individual monograph, and shake. After 15 minutes any opalescence produced is not more intense than that of a standard prepared in the same manner using a mixture of 10.0 ml of calcium standard solution (10 ppm Ca) and 5.0 ml of water in place of the test solution.
CHLORIDE
Dissolve the specified quantity of the substance in 30 to 40 ml of water, or, when the substance is already in solution, add water to make a total volume of 30 to 40 ml; or prepare a solution as directed in the monograph, and transfer to a comparison tube. Add 1 ml of nitric acid, except where nitric acid is used in the preparation of the solution. If, after acidification, the solution is not perfectly clear, filter it through a filter paper that gives negative test for chloride. Add 1 ml of silver nitrate TS, and sufficient water to make 50 ml. Mix, allow to stand for 5 minutes protected from direct sunlight. The opalescence produced is not greater than the standard opalescence, when viewed transversely.
Standard Opalescence
Measure the volume of 0.020 M hydrochloric acid, as directed in the individual monograph, into another comparison tube, dilute to a volume of 30 to 40 ml with water, add 1 ml of nitric acid, 1 ml of silver nitrate TS, and sufficient water to make 50 ml. Mix well, and allow to stand for 5 minutes.
FLUORIDE
(Note Use plasticware throughout this test.)
Reagent
pH 5.25 buffer Dissolve 110 g of sodium chloride and 1 g of sodium citrate dihydrate in 700 ml of water in a 2000-ml volumetric flask. Cautiously add 150 g of sodium hydroxide, and dissolve with shaking. Cool to room temperature, and while stirring, cautiously add 450 ml of glacial acetic acid to the cooled solution. Cool, add 600 ml of 2-propanol, dilute with water to volume, and mix: the pH of this solution is between 5.0 and 5.5.
Standard Stock Solution
Transfer 221 mg of sodium fluoride, previously dried at 150º for 4 hours, to a 100-ml volumetric flask, add 20 ml of water and mix to dissolve. Add 1.0 ml of 0.01 M sodium hydroxide, dilute with water to volume, and mix. Each ml of this solution contains 1 mg of fluoride ions. Store in a tightly closed, plastic container.
Standard Preparation
Dilute portions of the standard stock solution quantitatively and stepwise with pH 5.25 buffer to obtain 100-ml solutions having concentrations of 1, 3, 5, and 10 μg per ml.
Test Preparation
Unless otherwise specified in the monograph, prepare a test preparation as follows.
Transfer 1.0 g of the test substance, accurately weighed, to a 100-ml volumetric flask, dissolve in pH 5.25 buffer, dilute with pH 5.25 buffer to volume, and mix.
Procedure
Concomitantly measure the potential in millivolts, of the standard preparations and of the test preparation, with a pH meter capable of a minimum reproducibility of ±0.2 mV, equipped with a glass-sleeved calomelfluoride specific-ion electrode system. (Note When taking measurements, immerse the electrodes in the solution, which has been transferred to a 150-ml beaker containing a polytetrafluoroethylene-coated stirring bar. Allow to stir on a magnetic stirrer having an insulated top (or a thin asbestos sheet to prevent heat transfer) until equilibrium is attained (1 to 2 minutes), and record the potential. Rinse and dry the electrodes between measurements, being careful to avoid damaging the crystals of the specific-ion electrode.) Plot the logarithm of the fluoride-ion concentrations, in μg per ml, of the standard preparations versus the potential in millivolts. From the measured potential of the test preparation and the standard curve, determine the concentration, in μg per ml, of fluoride in the test preparation.
IRON
Reagent
Ammonium thiocyanate solution Dissolve 30 g of ammonium thiocyanate in water to make 100 ml.
Standard Preparation
Measure 1.0 ml of iron standard solution (10 ppm Fe) (Appendix 1.3) into a 50-ml comparison tube, add 40 ml of water and 2 ml of hydrochloric acid. Dilute with water to 50 ml, mix.
Test Preparation
Use the solution prepared as directed in the test for Iron in the individual monograph, and transfer it to a 50-ml comparison tube. Dilute with water to 50 ml, mix.
Procedure
To each of the tubes containing the standard preparation and the test preparation add 50 mg of ammonium peroxydisulfate crystals and 3 ml of ammonium thiocyanate solution, and mix, the colour of the solution from the test preparation is not darker than that of the solution from the standard preparation.
LEAD
The imposition of stringent limits on the amounts of lead that may be present in pharmaceutical products has resulted in the use of two methods, of which the one set forth following depends upon extraction of lead by solutions of dithizone. For determination of the content of heavy metals generally, expressed as a lead equivalent, see “Limit Test for Heavy Metals” (Appendix 5.2).
Select all reagents for this test to have as low a content of lead as practicable, and store all reagent solutions in containers of borosilicate glass. Rinse thoroughly all glassware with a warm 50 per cent v/v solution of nitric acid, followed by water.
Reagents
Ammonia-cyanide solution Dissolve 2 g of potassium cyanide in 15 ml of strong ammonia solution, and dilute with water to 10 ml.
Ammonium citrate solution Dissolve 40 g of citric acid in 90 ml of water. Add 2 or 3 drops of phenol red TS, then cautiously add strong ammonia solution until the solution acquires a reddish colour. Remove any lead that may be present by extracting the solution with 20-ml portions of dithizone extraction solution, until the dithizone solution retains its green colour.
Diluted lead standard solution Dilute an accurately measured volume of lead standard solution (10 ppm Pb) with 9 volumes of a 1 per cent v/v solution of nitric acid to obtain a solution that contains 1 μg of lead per ml.
Dithizone extraction solution Dissolve 30 mg of dithizone in 1000 ml of chloroform, and add 5 ml of ethanol. Store the solution in a refrigerator.
Before use, shake a suitable volume of the dithizone extraction solution with about half its volume of a 1 per cent v/v solution of nitric acid, discarding the nitric acid.
Hydroxylamine hydrochloride solution Dissolve 20 g of hydroxylamine hydrochloride in sufficient water to make approximately 65 ml. Transfer to a separator, add 5 drops of thymol blue TS, then add strong ammonia solution until the solution assumes a yellow colour. Add 10 ml of a 4 per cent w/v solution of sodium diethyldithiocarbamate, mix, and allow to stand for 5 minutes. Extract this solution with successive 10-to 15-ml portions of chloroform until a 5-ml portion of the chloroform extract does not assume a yellow colour when shaken with copper(II) sulfate TS. Add 3 M hydrochloric acid until the solution is pink (if necessary, add 1 or 2 drops more of thymol blue TS), and then dilute with water to 100 ml.
Potassium cyanide solution Dissolve 50 g of potassium cyanide in sufficient water to make 100 ml. Remove the lead from this solution by extraction with successive portions of dithizone extraction solution, as described under ammonium citrate solution above, then extract any dithizone remaining in the cyanide solution by shaking with chloroform, finally dilute the cyanide solution with sufficient water so that each 100 ml contains 10 g of potassium cyanide.
Standard dithizone solution Dissolve 10 mg of dithizone in 1000 ml of chloroform. Keep the solution in a glass-stoppered, lead-free bottle, suitably wrapped to protect it from light and store in a refrigerator.
Test Preparation
Unless otherwise specified in the monograph, prepare a test preparation as follows:
|
Caution Exercise safety precautions in this procedure, as some substances may react with explosive violence when digested with hydrogen peroxide. |
Transfer 1.0 g of the substance under test to a suitable flask, add 5 ml of sulfuric acid and a few glass beads, and digest on a hot plate in a hood until charring begins. Other suitable means of heating may be substituted. (Add additional sulfuric acid if necessary, to wet the substance completely, but do not add more than a total of 10 ml.) After the substance has been initially decomposed by the acid, add dropwise and with caution, strong hydrogen peroxide solution, allowing the reaction to subside and again heating between drops. Add the first few drops very slowly, mix carefully to prevent a rapid reaction, and discontinue heating if foaming becomes excessive. Swirl the solution in the flask to prevent unreacted substance from caking on the walls of the flask. (Note Add peroxide whenever the mixture turns brown or darkens.) Continue the digestion until the substance is completely destroyed, copious fumes of sulfur trioxide are evolved, and the solution is colourless. Cool, cautiously add 10 ml of water, evaporate until sulfur trioxide again is evolved, and cool.
Procedure
Transfer the test preparation or the volume of the prepared sample specified in the monograph to a separator and, unless otherwise directed in the monograph, add 6 ml of ammonium citrate solution and 2 ml of hydroxylamine hydrochloride solution. (For the determination of lead in iron salts use 10 ml of ammonium citrate solution.) Add 2 drops of phenol red TS, and make the solution just alkaline (red in colour) by the addition of strong ammonia solution. Cool the solution if necessary, and add 2 ml of potassium cyanide solution. Immediately extract the solution with 5-ml portions of dithizone extraction solution, draining off each extract into another separator, until the dithizone solution retains its green colour. Shake the combined dithizone solutions for 30 seconds with 20 ml of a 1 per cent w/v solution of nitric acid, and discard the chloroform layer. Add to the acid solution 5.0 ml of standard dithizone solution and 4 ml of ammonia-cyanide solution, and shake for 30 seconds: the colour of the chloroform layer is of no deeper shade of violet than that of a control made with a volume of diluted lead standard solution equivalent to the amount of lead permitted in the sample under examination, and with the same quantities of the same reagents and in the same manner as in the test with the sample.
SELENIUM
Reagent
Diaminonaphthalene solution Dissolve 100 mg of 2,3-diaminonaphthalene and 500 mg of hydroxylamine hydrochloride in 0.1 M hydrochloric acid to make 100 ml. Prepare this solution freshly on the day of use.
Standard Preparation
Pipette 6 ml of selenium standard solution (1 ppm Se) into a 150-ml beaker, and add 50 ml of a 1.6 per cent v/v solution of nitric acid.
Test Preparation
Using a 1000-ml combustion flask and using 25 ml of a 3.3 per cent v/v solution of nitric acid as the absorbing liquid, proceed as directed under “Oxygen Flask Combustion” (Appendix 6.2). Upon completion of the combustion, place a few ml of water in the cup, loosen the stopper, and rinse the stopper, the sample holder, and the sides of the flask with about 10 ml of water. Transfer the solution with the aid of about 20 ml of water to a 150-ml beaker, and heat gently to the boiling temperature. Boil for 10 minutes, and allow the solution to cool to room temperature.
Procedure
Treat the standard preparation, the test preparation, and the reagent blank consisting of 50 ml of a 1.6 per cent v/v solution of nitric acid, concomitantly and in parallel, as follows. Add a 50 per cent v/v solution of strong ammonia solution to adjust to a pH of 2.0±0.2. Dilute with water to 60.0 ml, and transfer to a low-actinic separator with the aid of 10.0 ml of water, adding the 10.0 ml of rinsings to the separator. Add 200 mg of hydroxylamine hydrochloride, swirl to dissolve, insert the stopper and swirl to mix. Allow the solution to stand at room temperature for 100 minutes. Add 5.0 ml of scyclohexane, shake vigorously for 2 minutes, and allow the layers to separate. Discard the aqueous layer, and centrifuge the cyclohexane extract to remove any traces of water. Determine the absorbance of each solution in a 1-cm cell at the wavelength of maximum absorbance at about 380 nm, with a suitable spectrophotometer, using cyclohexane as the blank, and compare the absorbances: the absorbance of the test preparation is not greater than of the standard preparation where a 200-mg test sample has been taken, or is not more than one-half that of the standard preparation where a 100-mg test sample has been taken.
SULFATE
Dissolve the specified quantity of the substance in 30 to 40 ml of water, or where the substance is already in solution, add water to make a total volume of 30 to 40 ml; or prepare a solution as directed in the monograph, and transfer to a comparison tube. Add 1 ml of dilute hydrochloric acid, except where hydrochloric acid is used in the preparation of the solution. If, after acidification, the solution is not perfectly clear, filter it through a filter paper that gives negative test for sulfate. Add 3 ml of barium chloride TS, and sufficient water to make 50 ml. Mix, allow to stand for 10 minutes. The turbidity produced is not more than the standard turbidity, when viewed transversely.
Standard Turbidity
Measure the volume of 0.010 M sulfuric acid, as directed in the individual monograph, into another comparison tube, dilute to a volume of 30 to 40 ml with water, add 1 ml of dilute hydrochloric acid, 3 ml of barium chloride TS, and sufficient water to make 50 ml. Mix well, and allow to stand for 10 minutes.